Caso del mes Abril 2017
« Todos los casos
Caso del mes Abril 2017
Descripción
Autores
- Dra. Juana Forner Giner
- Dr. Melchor Flores de la Torre
- Dr. Francisco Aparisi Rodriguez
Hospital Nueve de Octubre. Grupo NISA (Valencia)
Historia Clínica
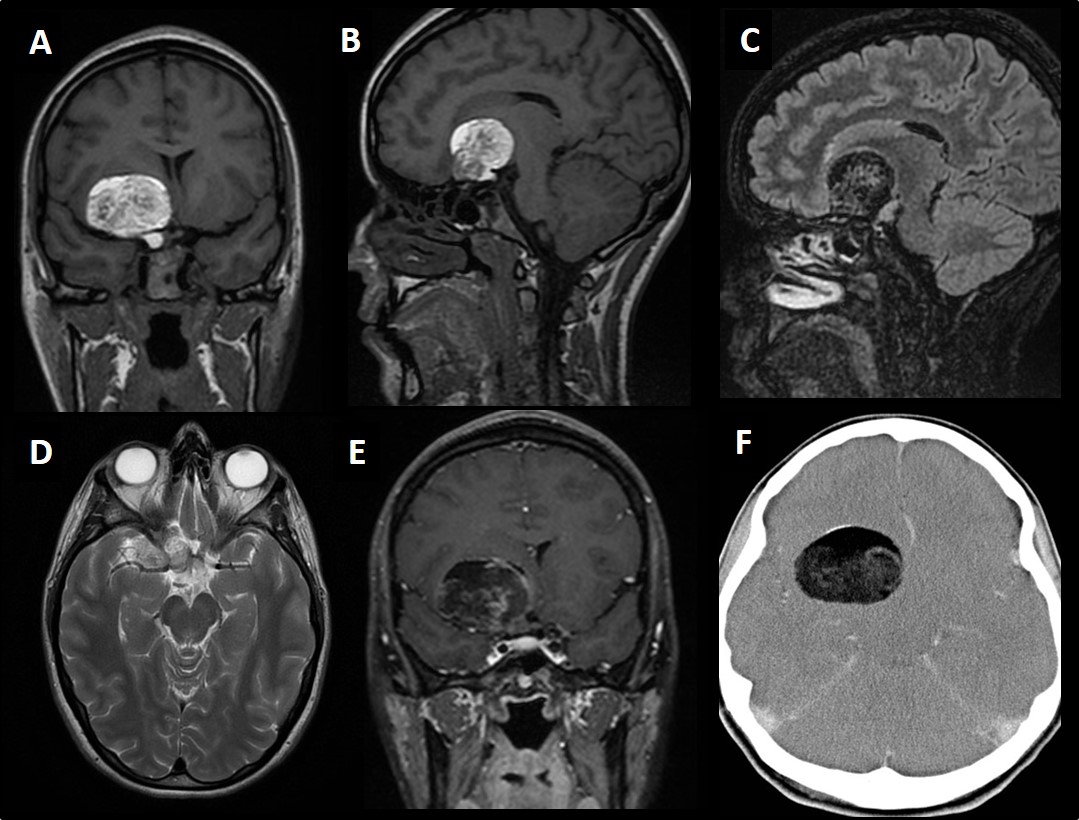
- Mujer de 50 años, con clínica de proptosis. Sin antecedentes conocidos ni otros síntomas.

Leyendas
Diagnóstico Anatomopatológico:
PSEUDOTUMOR ORBITARIO
RESUMEN:
El pseudotumor orbitario es la segunda causa más frecuente de proptosis. Consiste en un infiltrado inflamatorio no granulomatoso de localización variable en la órbita, que puede tener varias formas clínicas. Los pacientes pueden presentar quemosis, exoftalmos u oftalmoplejia dolorosa.
Su causa es inflamatoria y generalmente unilateral. Existen múltiples patrones radiológicos, en este caso se trata de una miositis ( que es el patrón más frecuente). Suele afectar a uno o varios músculos, sobre todo rectos inferiores y laterales. Como hallazgo típico puede observarse el engrosamiento de la inserción anterior del músculo y de la úvea-esclera adyacente. Se presenta hipointensa en las secuencias T1, y T2, con intenso realce con Gadolinio. Puede afectar al espacio intra y extraconal (como es este caso) con formas infiltrativas difusas que pueden extenderse fuera de la órbita y afectar al ápex petroso , fosa pterigo-palatina y seno cavernoso.
La afectación incluye la inserción tendinosa anterior del músculo (a diferencia de la oftalmopatia tiroidea)
El tratamiento son los corticoides.
DIAGNÓSTICO DIFERENCIAL:
– Oftalmopatía tiroidea (enfermedad de Graves) : se observa engrosamiento de la musculatura extrínseca ocular, respetando las inserciones tendinosas.
– Patología infecciosa e inflamatoria.
– Linfoma: lesión tumoral maligna más frecuente en esta topografía. Captación homogénea.
– Metástasis.
BIBLIOGRAFÍA:
– Inflammatory pseudotumor orbitae: an atypical manifestation of giant cell arteritis. Hittinger M, Berlis A, Pfadenhauer K. Clin Neuroradiol. 2015 Dec;25(4):411-4″
Diagnóstico
DIAGNÓSTICO
Encefalopatía posterior reversible durante el puerperio secundaria a eclampsia.
Es frecuente encontrarlo en mujeres jóvenes.
La clínica neurológica aguda es variable: encefalopatía, convulsiones, cefalea, y menos frecuente alteraciones visuales y status.
En el 80% de los casos está relacionado con la hipertensión.
Fisiopatológicamente traduce un edema vasogénico, que suele verse en la TC como hipodensidad en la sustancia blanca subcortical bilateral asimétrica predominantemente en región parieto-occipital. Puede haber otros patrones como del surco frontal superior, holohemisférico, u otros menos frecuentes donde hay afectación del cerebelo, ganglios basales y la médula. Generalmente, hay combinación de patrones.
No siempre es reversible y no siempre es posterior.
Puede progresar a hemorragia en forma HSA o un hematoma parenquimatoso en 10-25% de los casos.
Tratamiento: Tratamiento y profilaxis de nuevos episodios de eclampsia con sulfato de magnesio. Control estricto de cifras tensionales. Vigilancia de nivel de conciencia. Control analítico seriado.
Diagnóstico diferencial:
· Leucoencefalopatía multifocal progresiva (LMP)
· Encefalopatía hipoglucémica
· Infarto de la circulación posterior o múltiples territorios
· Gliomatosis cerebri
· Trombosis del seno sagital
· Encefalopatía hipóxico-isquémica
· Angiopatía amiloide cerebral inflamatoria
BIBLIOGRAFÍA
Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008 Feb;65(2):205-10. doi: 10.1001/archneurol.2007.46. PMID: 18268188.
Fugate JE, Rabinstein AA. Posterior reversible encephalopathy syndrome: clinical and radiological manifestations, pathophysiology, and outstanding questions. Lancet Neurol. 2015 Sep;14(9):914-925. doi: 10.1016/S1474-4422(15)00111-8. Epub 2015 Jul 13. Erratum in: Lancet Neurol. 2015 Sep;14(9):874. PMID: 26184985.
McKinney AM, Short J, Truwit CL, McKinney ZJ, Kozak OS, SantaCruz KS, et al. Posterior reversible encephalopathy syndrome: incidence of atypical regions of involvement and imaging findings. AJR Am J Roentgenol. (2007) 189:904–12. doi: 10.2214/AJR.07.2024